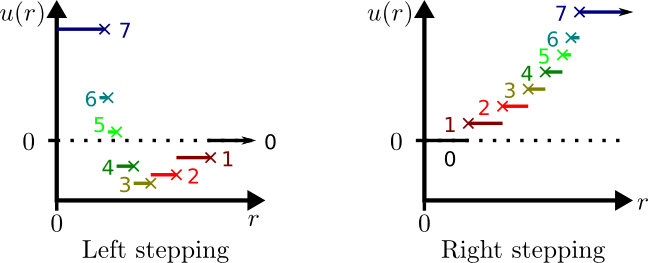This is a reference for the configuration file format of DynamO. If
you are new to DynamO, please see
the general introduction to the file
format in tutorial 3 first.
In the following sections, all of the available XML tags and
attributes of the configuration file are listed and all of the
options are detailed for each tag type. Below is a hyperlinked
hierarchy of the tags in the configuration file.
General Structure
-
DynamOconfig
-
Simulation1
-
Properties1
-
ParticleData1
1These tags are just containers and do not have any
attributes.
Scheduler
The Scheduler specifies how DynamO searches the simulation for
events. How the events are actually sorted by the scheduler is
specified through the Sorter tag.
Type="Dumb"
Description
The "Dumb" scheduler is the most basic and
slowest scheduler available. When particles undergo an event, the
Dumb scheduler tests for new events against all other particles in
the system (regardless of where they are). This cost scales linearly
with the system size ($\mathcal{O}(N)$), resulting in an overall
$\mathcal{O}(N^2)$ scaling of the computational cost. This Scheduler
type is only provided for debugging and testing purposes.
Example usage
<Scheduler Type="Dumb">
<Sorter .../>
</Scheduler>
Full Tag, Subtag, and Attribute List
-
Type (attribute): Must have the
value "Dumb" to select this Scheduler type.
-
Sorter (tag): This tag specifies the type of event
sorter used in the Scheduler. See the section
on Sorters for more information on this tag.
Type="NeighbourList"
Description
The "NeighbourList" scheduler uses a
NeighbourList to optimise the detection of events. When particles
undergo an event, the NeighbourList scheduler only tests for new
events against nearby particles. This cost is independent of the
system size ($\mathcal{O}(1)$), resulting in an overall linear
($\mathcal{O}(N)$) scaling of the computational cost.
Note
The NeighbourList scheduler will automatically add
a Global neighbour-list interaction to the
system if one has not already been specified. The current default is
the Cells
type Global.
If you want to specify the neighbour list type or change its
parameters, please add a Global with the name
attribute set to "SchedulerNBList" to allow the NeighbourList
Scheduler to identify it.
Example usage
<Scheduler Type="NeighbourList">
<Sorter .../>
</Scheduler>
Full Tag, Subtag, and Attribute List
-
Type (attribute): Must have the
value "NeighbourList" to select this Scheduler type.
-
Sorter (tag): This tag specifies the type of event
sorter used in the Scheduler. See the section
on Sorters for more information on this tag.
Sorter
The Sorter tag specifies the method DynamO uses to sort events when
determining the next event to occur.
Type="CBT"
Description
The "CBT" Sorter uses a STL priority queue for
each particle and inserts this into a Complete Binary Tree (CBT) to
sort the events. This type of Sorter is very robust to unusual
systems (such as systems with zero or one particle) but, as the
computational cost scales as $\mathcal{O}(\log_2(N)$ with the system
size, it is not the default Sorter used by DynamO.
Example usage
<Sorter Type="CBT"/>
Full Tag, Subtag, and Attribute List
-
Type (attribute): Must have the
value "CBT" to select this Sorter type.
Type="BoundedPQMinMax3"
Description
The "BoundedPQMinMax3" Sorter uses a bounded
MinMax heap of size 3 to sort particle events. These particle queues
are then presorted using a bounded priority queue. The earliest
entry in the bounded priority queue is then sorted using a Complete
Binary Tree. In this way, the lazy deletion scheme can be combined
with a fixed size event queue and a bounded priority queue to yield
a constant ($\mathcal{O}(1)$) scaling of the computational cost with
the system size. There are variants of this scheduler with different
sizes of the MinMax heaps ranging from 2 to 8 (e.g.,
"BoundedPQMinMax8" is also available). After many years of testing
this has proven to be the fastest and lowest memory event sorter for
a range of event-driven particle simulations. In small systems the
CBT Sorter is slightly faster and, depending on the system studied,
you may find the MinMax heap size might be increased or decreased to
increase performance.
Example usage
<Sorter Type="BoundedPQMinMax3"/>
Full Tag, Subtag, and Attribute List
-
Type (attribute): Must have the
value "BoundedPQMinMax3" to select this Sorter
type. Other variants from "BoundedPQMinMax2" to
"BoundedPQMinMax8" are also available.
SimulationSize
Description
The SimulationSize tag specifies the dimensions
of the primary image for periodic boundary conditions. When the
system is not periodic, it specifies the size of the tiled
neighbourlist (if one is used). If no neighbour list is used in an
infinite system, this tag has no effect.
Example usage
This example specifies a $10\times10\times10$ primary image.
<SimulationSize x="10" y="10" z="10"/>
Full Tag, Subtag, and Attribute List
-
x (attribute): The size in the $x$ dimension.
-
y (attribute): The size in the $y$ dimension.
-
z (attribute): The size in the $z$ dimension.
Species
Species are vital tags used to specify the mass and inertia data of
a set of particles. They also provide a unique identifier/name for
groups of particles as each particle must belong to exactly one
species. Many output plugins use the species of a particle to
separate results (for example, a radial distribution function will
be generated for all pairings of species in the system).
Particles will have rotational degrees of freedom if they have a
Species which provides a non-zero inertia tensor
(e.g. "SphericalTop"). Other Species
types such as "Point"
and "FixedCollider" will avoid the
computational overhead of tracking the rotation of the particles.
Type="Point"
Description
This Species type corresponds to point mass
(zero inertia) particles, but this type is also used in systems
where inertial data is unimportant (atomic or frictionless
systems). It is the simplest type of Species available.
Example usage
<Species Mass="1" Name="Bulk" Type="Point">
<IDRange .../>
</Species>
Full Tag, Subtag, and Attribute List
-
Type (attribute): Must have the
value "Point" to select this Species type.
-
Mass (attribute): The mass of the particles
represented by this Species.
This attribute is a Property specifier with units
of Mass (see the section on Properties
for more information).
-
Name (attribute): The name of the particles
represented by this Species. This is used in output, so species
"A" or "Carbon" are examples. If the system is monocomponent,
dynamod often uses the name "Bulk".
-
IDRange (tag): A IDRange which specifies the
Particles represented by this Species tag. The IDRanges of each
Species must not overlap with any other Species in the
system. All particles must belong to exactly one Species.
Type="FixedCollider"
Description
This Species type corresponds to particles which
have infinite mass and no inertia tensor. This is useful for
particles which are used as the boundaries of a system (also called
a particle mesh).
Example usage
<Species Name="Bulk" Type="FixedCollider">
<IDRange .../>
</Species>
Full Tag, Subtag, and Attribute List
-
Type (attribute): Must have the
value "FixedCollider" to select this Species type.
-
Name (attribute): The name of the particles
represented by this Species. This is used in output, so species
"A" or "Carbon" are examples. If the system is monocomponent,
dynamod often uses the name "Bulk".
-
IDRange (tag): A IDRange which specifies the
Particles represented by this Species tag. The IDRanges of each
Species must not overlap with any other Species in the
system. All particles must belong to exactly one Species.
Type="SphericalTop"
Description
This Species type corresponds to particles where
the three principal momenta of inertia are identical. It is also
used in systems where only two of the principal momenta of inertia
are equal but the rotation is constrained such that the particle
cannot precess.
Example usage
<Species Mass="1" Name="Bulk" Type="SphericalTop" InertiaConstant="0.1">
<IDRange .../>
</Species>
Full Tag, Subtag, and Attribute List
-
Type (attribute): Must have the
value "SphericalTop" to select this Species type.
-
Mass (attribute): The mass of the particles
represented by this Species.
This attribute is a Property specifier with units
of Mass (see the section on
Properties for more information).
-
InertiaConstant (attribute): The area factor of the
principal momenta of inertia of the particles represented by this
Species. This is value is multiplied by the mass of the particle
to obtain the value of the principal momenta of inertia.For a
solid sphere this value should be $\sigma^2/10$ where $\sigma$ is
the particle
diameter. A
list of inertia constants is available at wikipedia.
-
Name (attribute): The name of the particles
represented by this Species. This is used in output, so species
"A" or "Carbon" are examples. If the system is monocomponent,
dynamod often uses the name "Bulk".
-
IDRange (tag): A IDRange which specifies the
Particles represented by this Species tag. The IDRanges of each
Species must not overlap with any other Species in the
system. All particles must belong to exactly one Species.
BC (Boundary Conditions)
The BC tag in the configuration file controls the boundary
conditions of the simulation.
Type="None"
Description
The "None" boundary condition actually
corresponds to an infinite system, without boundaries. The positions
of the particles are not restricted in any dimension.
Example usage
<BC Type="None"/>
Full Tag, Subtag, and Attribute List
-
Type (attribute): Must have the
value "None" to select this BC type.
Type="PBC"
Description
The "PBC" boundary condition applies periodic
boundary conditions to every dimension. The positions of the
particles are wrapped to fit within the primary image, whose
dimensions are specified by
the SimulationSize tag.
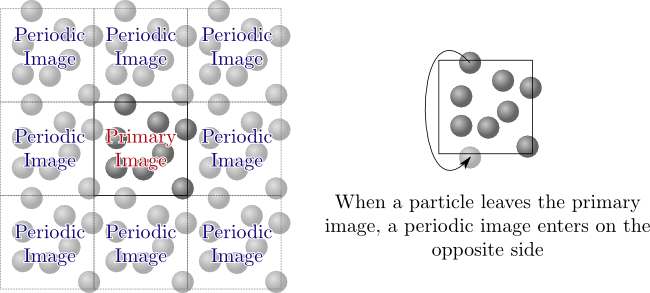
When we want to study molecular fluids we often want the "bulk"
properties of the fluid. Effects like surface tension will have a
strong influence if there are any free surfaces or boundaries in
contact with the fluid, as systems simulated are usually relatively
small ($\approx10^5$ molecules). On the other hand, there must be
some boundary used to contain the fluid, as using an open (infinite
size) system will cause the fluid to either evaporate or form
droplets, again with surface effects. To avoid the effects of
boundaries/walls while still "containing" the
system, periodic
boundary conditions are often used. With periodic boundaries, a
small representative amount of fluid, called the "primary image," is
simulated. This primary image is then surrounded with periodic
images which are copies of the primary image as illustrated in the
figure below:
These boundaries allow the approximation of an infinite fluid using
a small repeating image. This is an approximation as the periodicity
adds additional correlations to the system, but it is a convenient
technique to avoid using real boundaries such as walls to contain
the system. When using periodic boundary conditions it is still
possible to enter into two-phases if the simulation has attractive
interactions, so care must still be taken.
Example usage
<BC Type="PBC"/>
Full Tag, Subtag, and Attribute List
-
Type (attribute): Must have the
value "PBC" to select this BC type.
Type="LE"
Description
The "LE" boundary condition applies
Lees-Edwards boundary conditions to the system. These are periodic
boundary conditions but they shear the system by setting the
periodic images in the $y$ direction in motion in the $x$ direction
(see figure below). They are also known as sliding-brick boundary
conditions.
Example usage
<BC Type="LE" DXD="0" Rate="1"/>
Full Tag, Subtag, and Attribute List
-
Type (attribute): Must have the
value "LE" to select this BC type.
-
DXD (attribute): This attribute stores the current
displacement of the nearest periodic images in the $x$-direction
(units of length).
-
Rate (attribute): This specifies how fast the
boundary is shearing (units of primary image length per unit
time).
Structure
Structure tags are used to specify multi-particle structures, such
as molecules. This is to mark these structures to allow specialized
data collection and to simplify the implementation of complex
interactions.
Each Structure tag contains one or
more IDRange tags, each of which specifies a
single structure of the same type. So one Structure tag has at least
the following basic format:
<Topology ...>
<Structure ...>
<IDRange .../>
<IDRange .../>
...
</Structure>
</Topology>
Type="Chain"
Description
The "Chain" Structure is used to mark out a single linear chain
molecule.
Example usage
<Structure Type="Chain" Name="Polymer">
<IDPairRange .../>
</Interaction>
Full Tag, Subtag, and Attribute List
-
Type (attribute): Must have the
value "Chain" to select this Interaction type.
-
Name (attribute): The name of the Structure. This
name is used to identify the Structure in the output generated by
the dynarun command. Each Structure should have a name which is
unique.
-
IDRange (tag):
This IDRange tag specifies the particles
which belong to the linear chain. See
the section on IDRange tags for more
information on the format of this tag.
Interaction
Interaction tags are used to specify how pairs of particles interact
and generate Interaction events.
Each pair of Particle ID's must have a corresponding
Interaction.
Every possible pairing of particles
(including self pairings)
must have a corresponding Interaction, even if they don't
interact. If you don't want them to interact at all, you must use a
Null Interaction.
The order in which Interactions are listed in the configuration
file is important.
When DynamO tests for
interactions/events between a pair of particles, it moves through
the list of interactions in the order in which they are specified,
testing if the ID's of the pair match the
Interaction's IDPairRange. Interactions
which are higher in the configuration file will override matching
Interactions which are lower down.
Each particle must also have a self-Interaction.
This
self-Interaction does not generate events, but is used to decide how
to draw the particle in the visualiser and to calculate some single
particle properties, such as the excluded volume.
Type="Null"
Description
The "Null" Interaction is used to mark particle
pairs out as non-interacting. All particle pairs must have a
corresponding Interaction defined, so this Interaction is the only
way to prevent events being generated for a set of particle pairs.
Example usage
<Interaction Type="Null" Name="Bulk">
<IDPairRange .../>
</Interaction>
Full Tag, Subtag, and Attribute List
-
Type (attribute): Must have the
value "Null" to select this Interaction type.
-
Name (attribute): The name of the
Interaction. This name is used to identify the Interaction in
the configuration file (e.g.,
see Species) and in the output generated
by the dynarun command. Each Interaction must have a name which
is unique.
-
IDPairRange (tag): This IDPairRange tag specifies
the pairs of particles which interact using this
Interaction. See the section on
IDPairRanges for more information on the format of this tag.
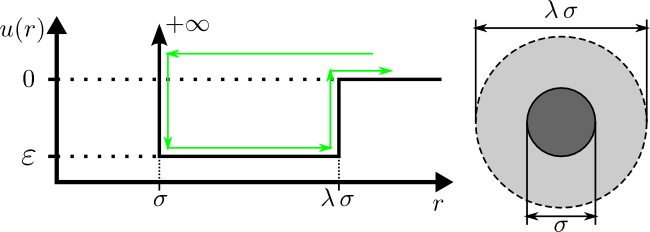
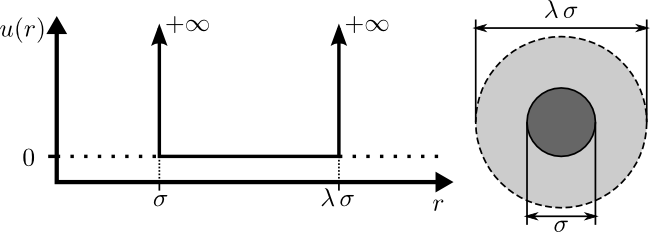
Type="HardSphere"
Description
The "HardSphere" Interaction implements the
hard-sphere interaction potential. This is one of the simplest
event-driven potentials available.
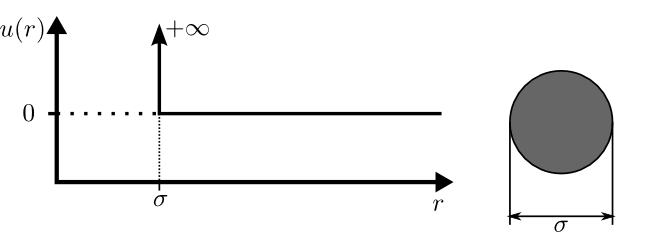
A hard sphere is a simple molecular model used to capture the
fundamental effects of "excluded-volume" interactions. You may think
of the hard-sphere fluid as an extension of the ideal-gas model,
where each molecule now has a diameter, $\sigma$, and cannot overlap
the volume of this diameter with the volume of other molecules. The
parameters of this model are illustrated below:
where $u(r)$ is the interparticle potential (which is the potential
energy between two particles separated by a distance of
$r$). Particles do not interact at separations greater than the
diameter of the molecule ($u(r)=0$ for $r\in[\sigma,\,\infty]$). The
infinite interaction energy of the hard core ($u(r)=+\infty$ for
$r\in[0,\,\sigma]$) makes it energetically impossible for particles
to "overlap", therefore the particles will elastically bounce-off of
each other when they come into contact.
The effects of this additional "excluded volume" interaction is
dramatic and leads to complex transport coefficients as a function of
density and the appearance of a fluid-solid freezing transition. This
model is too simple to capture any complex temperature effects, such
as a liquid/gas phase transition, as it has no finite interaction
energies (unlike the square-well
fluid). Despite its simplicity, the structure of many real
crystals is dominated by the repulsive "excluded-volume" interactions
caused by overlapping electron clouds which may be effectively
captured by the hard-sphere model. It is also at the heart of kinetic
theory which is the most successful attempt to predict the transport
properties of fluids from their molecular interactions. The
interparticle potential of this model is given in the figure below:
Collision rule
To perform an interaction we need a collision rule which calculates
the post-collision velocities of the two particles undergoing the
Interaction. The collision rule expresses the post-collision
velocities in terms of the pre-collision values.
Show/hide derivation of collision rule
Using the definition of the relative velocity
$\boldsymbol{v}_{ij}=\boldsymbol{v}_{i}-\boldsymbol{v}_{j}$ of two
particles $i$ and $j$, and the identities
$\boldsymbol{v}_i=\boldsymbol{v}_{ij}+\boldsymbol{v}_j$ and
$\boldsymbol{v}_j=\boldsymbol{v}_i-\boldsymbol{v}_{ij}$, we have
\[\begin{align}
\boldsymbol{v}_j'-\boldsymbol{v}_j&=\boldsymbol{v}_i'-\boldsymbol{v}_i - \boldsymbol{v}_{ij}'+\boldsymbol{v}_{ij}
\end{align}\]
where the primes denote post-collision values. Using the conservation
of momentum, we can write
\[\begin{align}
m_i\,\boldsymbol{v}_i'+m_j\,\boldsymbol{v}_j'&=m_i\,\boldsymbol{v}_i+m_j\,\boldsymbol{v}_j\\
\boldsymbol{v}_i'-\boldsymbol{v}_i&=-\frac{m_j}{m_i}\left(\boldsymbol{v}_j'-\boldsymbol{v}_j\right)
\end{align}\]
where $m_i$ is the mass of particle $i$. Using the equation
derived from the conservation of momentum to eliminate
$\boldsymbol{v}_j$ terms, we have
\[\begin{align}
\boldsymbol{v}_i'-\boldsymbol{v}_i&=-\frac{m_j}{m_i}\left(\boldsymbol{v}_i'-\boldsymbol{v}_i - \boldsymbol{v}_{ij}'+\boldsymbol{v}_{ij}\right)\\
&=m_i^{-1}\,\mu\left(\boldsymbol{v}_{ij}'-\boldsymbol{v}_{ij}\right)
\end{align}\]
where $\mu_{ij}=\left(m_i^{-1}+m_j^{-1}\right)^{-1}$ is the reduced
mass. Therefore we can calculate the post-collision velocities of the
particles if we know the change in the relative velocities. For smooth
particles the velocities only change along the line of contact, and we
have:
\[\begin{align*}
\left[\boldsymbol{v}_{ij}'\right]_\parallel&=-\varepsilon\left[\boldsymbol{v}_{ij}\right]_\parallel &
\left[\boldsymbol{v}_{ij}'\right]_\perp&=\left[\boldsymbol{v}_{ij}\right]_\perp
\end{align*}\]
where $\varepsilon$ is the elasticity/coefficient of restitution
and the subscript $\parallel$ and $\perp$ denote the components
parallel and perpendicular to the line of contact. These are
calculated like so
$\boldsymbol{v}_{ij,\parallel}=\hat{\boldsymbol{r}}_{ij}\left(\hat{\boldsymbol{r}}_{ij}\cdot\boldsymbol{v}_{ij}\right)$
and
$\boldsymbol{v}_{ij,\perp}=-\hat{\boldsymbol{r}}_{ij}\times\left(\hat{\boldsymbol{r}}_{ij}\times\boldsymbol{v}_{ij}\right)$
where $\hat{\boldsymbol{r}}_{ij}$ is the unit vector in the
direction of the relative separation at contact,
$\boldsymbol{r}_{ij}=\boldsymbol{r}_i-\boldsymbol{r}_j$, and
$\boldsymbol{r}_i$ is the position of particle $i$. Combining
these rules results in the following expression
\[\begin{align*}
\boldsymbol{v}_{ij}'-\boldsymbol{v}_{ij}=-(1+\varepsilon)\left(\hat{\boldsymbol{r}}_{ij}\cdot\boldsymbol{v}_{ij}\right)\hat{\boldsymbol{r}}_{ij}
\end{align*}\]
This leads to the final expressions for smooth particles:
\[\begin{align}
\boldsymbol{v}_i'-\boldsymbol{v}_i&=-m_i^{-1}\,\mu_{ij}(1+\varepsilon)\left(\hat{\boldsymbol{r}}_{ij}\cdot\boldsymbol{v}_{ij}\right)\hat{\boldsymbol{r}}_{ij}\\
\boldsymbol{v}_j'-\boldsymbol{v}_j&=+m_j^{-1}\,\mu_{ij}(1+\varepsilon)\left(\hat{\boldsymbol{r}}_{ij}\cdot\boldsymbol{v}_{ij}\right)\hat{\boldsymbol{r}}_{ij}
\end{align}\]
In rough systems, particles will change rotation due to
interactions. To define the dynamics in such a system we need to
consider the relative surface velocity at the point of contact,
$\boldsymbol{g}_{ij}$, calculated as follows \[\begin{align*}
\boldsymbol{g}_{ij}&=\left(\boldsymbol{v}_i-\boldsymbol{\omega}_i\times
R_i\,\hat{\boldsymbol{r}}_{ij}\right)-\left(\boldsymbol{v}_j+\boldsymbol{\omega}_j\times
R_j\hat{\boldsymbol{r}}_{ij}\right)\\
&=\boldsymbol{v}_{ij}-\left(R_i\,\boldsymbol{\omega}_i+R_j\,\boldsymbol{\omega}_j\right)\times\hat{\boldsymbol{r}}_{ij}
\end{align*}\] where $\boldsymbol{\omega}_j$ is the angular velocity
and $R_i$ is the radus of particle $i$. In this case, we can define a
normal and a tangential coefficient of restitution as follows
\[\begin{align}
\left[\boldsymbol{g}_{ij}\right]_\parallel'&=-\varepsilon^n\left[\boldsymbol{g}_{ij}\right]_\parallel
&
\left[\boldsymbol{g}_{ij}\right]_\perp'&=\varepsilon^t\left[\boldsymbol{g}_{ij}\right]_\perp
\end{align}\]
where
$\boldsymbol{g}_{ij,\parallel}=\hat{\boldsymbol{r}}_{ij}\left(\hat{\boldsymbol{r}}_{ij}\cdot\boldsymbol{g}_{ij}\right)$
and
$\boldsymbol{g}_{ij,\perp}=-\hat{\boldsymbol{r}}_{ij}\times\left(\hat{\boldsymbol{r}}_{ij}\times\boldsymbol{g}_{ij}\right)$. Noting
that
$\boldsymbol{x}=\hat{\boldsymbol{e}}(\hat{\boldsymbol{e}}\cdot\boldsymbol{x})-\hat{\boldsymbol{e}}\times\left(\hat{\boldsymbol{e}}\times\boldsymbol{x}\right)$,
these definitions lead to the following expressions
\[\begin{align*}
\hat{\boldsymbol{r}}_{ij}\left(\hat{\boldsymbol{r}}_{ij}\cdot\left[\boldsymbol{g}_{ij}'-\boldsymbol{g}_{ij}\right]\right)&=-(1+\varepsilon^n)\hat{\boldsymbol{r}}_{ij}\left(\hat{\boldsymbol{r}}_{ij}\cdot\boldsymbol{g}_{ij}\right)
&
\hat{\boldsymbol{r}}_{ij}\times\left(\hat{\boldsymbol{r}}_{ij}\times\left[\boldsymbol{g}_{ij}'-\boldsymbol{g}_{ij}\right]\right)&=(\varepsilon^t-1)\hat{\boldsymbol{r}}_{ij}\times\left(\hat{\boldsymbol{r}}_{ij}\times\boldsymbol{g}_{ij}\right)
\end{align*}\]
We need to find an expression which closes the linear momentum balance
by providing a relationship between $\boldsymbol{v}_{ij}'-\boldsymbol{v}_{ij}$ and
$\boldsymbol{g}_{ij}'-\boldsymbol{g}_{ij}$, as well as determining how the angular velocity
changes. Beginning with the angular velocity, we take the contact
point as the origin about which the angular momentum is defined. We
then note that all contact forces act through the origin and see that
angular velocity is conserved for both particles separately. Thus we
will have two separate angular-momentum conservation rules:
\[\begin{align*}
m_i\,R_i\,\hat{\boldsymbol{r}}_{ij}\times\boldsymbol{v}_i' + \boldsymbol{I}_i\cdot\boldsymbol{\omega}_i' &= m_i\,R_i\,\hat{\boldsymbol{r}}_{ij}\times\boldsymbol{v}_i + \boldsymbol{I}_i\cdot\boldsymbol{\omega}_i\\
m_j\,R_j\,\hat{\boldsymbol{r}}_{ij}\times\boldsymbol{v}_j' - \boldsymbol{I}_j\cdot\boldsymbol{\omega}_j' &= m_j\,R_j\,\hat{\boldsymbol{r}}_{ij}\times\boldsymbol{v}_j - \boldsymbol{I}_j\cdot\boldsymbol{\omega}_j
\end{align*}\]
For symmetric objects we have an isotropic inertia tensor
$\boldsymbol{I}_i=I_i\,\boldsymbol{1}$, and can define a reduced moment of inertia as
$\tilde{I}_i=I_i/m_i\,R_i^2$ and can rearrange these two equations
like so
\[\begin{align*}
\boldsymbol{\omega}_i'- \boldsymbol{\omega}_i&=- R_i^{-1}\tilde{I}_i^{-1}\left[\hat{\boldsymbol{r}}_{ij}\times\left(\boldsymbol{v}_i' -\boldsymbol{v}_i\right)\right]\\
\boldsymbol{\omega}_j'- \boldsymbol{\omega}_j &= + R_j^{-1}\tilde{I}_j^{-1}\left[\hat{\boldsymbol{r}}_{ij}\times\left(\boldsymbol{v}_j' -\boldsymbol{v}_j\right)\right]
\end{align*}\]
We need to find the post-collision linear velocities to close these
expressions. We can define the pre and post collision relative
velocity at the surface as follows
\[\begin{align*}
\boldsymbol{g}_{ij}&=\left(\boldsymbol{v}_i-\boldsymbol{\omega}_i\times
R_i\,\hat{\boldsymbol{r}}_{ij}\right)-\left(\boldsymbol{v}_j+\boldsymbol{\omega}_j\times
R_j\hat{\boldsymbol{r}}_{ij}\right)\\
&=\boldsymbol{v}_{ij}-\left(R_i\,\boldsymbol{\omega}_i+R_j\,\boldsymbol{\omega}_j\right)\times\hat{\boldsymbol{r}}_{ij}
\end{align*}\]
Finding the post collision value of this
\[\begin{align*}
\boldsymbol{g}_{ij}'&=\boldsymbol{v}_{ij}'-\left(R_i\,\boldsymbol{\omega}_i'+R_j\,\boldsymbol{\omega}_j'\right)\times\hat{\boldsymbol{r}}_{ij}\\
&=\boldsymbol{v}_{ij}' - \left(R_i\left(\boldsymbol{\omega}_i - R_i^{-1}\,\tilde{I}_i^{-1}\left[\hat{\boldsymbol{r}}_{ij}\times\left(\boldsymbol{v}_i' -\boldsymbol{v}_i\right)\right]\right)+R_j\left(\boldsymbol{\omega}_j + R_j^{-1}\,\tilde{I}_j^{-1}\left[\hat{\boldsymbol{r}}_{ij}\times\left(\boldsymbol{v}_j' -\boldsymbol{v}_j\right)\right]\right)\right)\times\hat{\boldsymbol{r}}_{ij}\\
&=\boldsymbol{v}_{ij}'-\left(R_i\,\boldsymbol{\omega}_i +
R_j\,\boldsymbol{\omega}_j\right)\times\hat{\boldsymbol{r}}_{ij} +
\left(\tilde{I}_i^{-1}\left[\hat{\boldsymbol{r}}_{ij}\times\left(\boldsymbol{v}_i'
-\boldsymbol{v}_i\right)\right]
-\tilde{I}_j^{-1}\left[\hat{\boldsymbol{r}}_{ij}\times\left(\boldsymbol{v}_j'
-\boldsymbol{v}_j\right)\right]\right)\times\hat{\boldsymbol{r}}_{ij}
\end{align*}\]
Subtracting the pre-collision value from this value, and assuming that
all particles have the same reduced moment of inertia, we have
\[\begin{align*}
\boldsymbol{g}_{ij}'-\boldsymbol{g}_{ij}&=\boldsymbol{v}_{ij}'-\boldsymbol{v}_{ij}+
\tilde{I}^{-1}\left(\left[\hat{\boldsymbol{r}}_{ij}\times\left(\boldsymbol{v}_i'
-\boldsymbol{v}_i\right)\right]
-\left[\hat{\boldsymbol{r}}_{ij}\times\left(\boldsymbol{v}_j'
-\boldsymbol{v}_j\right)\right]\right)\times\hat{\boldsymbol{r}}_{ij}\\
&=\boldsymbol{v}_{ij}'-\boldsymbol{v}_{ij}-
\tilde{I}^{-1}\,\hat{\boldsymbol{r}}_{ij}\times\left[\hat{\boldsymbol{r}}_{ij}\times\left(\boldsymbol{v}_{ij}'
-\boldsymbol{v}_{ij}\right)\right]
\end{align*}\]
Now that we have the relationship between $\boldsymbol{g}_{ij}'-\boldsymbol{g}_{ij}$
and $\boldsymbol{v}_{ij}'-\boldsymbol{v}_{ij}$, we look to insert the coefficients of
restitution. We notice that the second term is perpendicular to
$\hat{\boldsymbol{r}}_{ij}$, therefore we have
\[\begin{align}
\hat{\boldsymbol{r}}_{ij}\cdot\left(\boldsymbol{v}_{ij}'-\boldsymbol{v}_{ij}\right) = \hat{\boldsymbol{r}}_{ij}\cdot\left(\boldsymbol{g}_{ij}'-\boldsymbol{g}_{ij}\right)
\end{align}\]
Taking the vector product instead and we have
\[\begin{align*}
\hat{\boldsymbol{r}}_{ij}\times\left[\boldsymbol{g}_{ij}'-\boldsymbol{g}_{ij}\right] &=\hat{\boldsymbol{r}}_{ij}\times\left[\boldsymbol{v}_{ij}'-\boldsymbol{v}_{ij}\right]-
\tilde{I}^{-1}\hat{\boldsymbol{r}}_{ij}\times\left[\hat{\boldsymbol{r}}_{ij}\times\left[\hat{\boldsymbol{r}}_{ij}\times\left(\boldsymbol{v}_{ij}'
-\boldsymbol{v}_{ij}\right)\right]\right]
\end{align*}\]
we note that
$\hat{\boldsymbol{e}}\times\left(\hat{\boldsymbol{e}}\times\left(\hat{\boldsymbol{e}}\times\boldsymbol{x}\right)\right)=-\hat{\boldsymbol{e}}\times\boldsymbol{x}$,
giving
\[\begin{align}
\hat{\boldsymbol{r}}_{ij}\times\left[\boldsymbol{g}_{ij}'-\boldsymbol{g}_{ij}\right]
&=\hat{\boldsymbol{r}}_{ij}\times\left[\boldsymbol{v}_{ij}'-\boldsymbol{v}_{ij}\right]+
\tilde{I}^{-1}\,\hat{\boldsymbol{r}}_{ij}\times\left(\boldsymbol{v}_{ij}'
-\boldsymbol{v}_{ij}\right)\nonumber\\
\hat{\boldsymbol{r}}_{ij}\times\left[\boldsymbol{v}_{ij}'-\boldsymbol{v}_{ij}\right]&=\left(1+\tilde{I}^{-1}\right)^{-1}\hat{\boldsymbol{r}}_{ij}\times\left[\boldsymbol{g}_{ij}'-\boldsymbol{g}_{ij}\right]
\end{align}\]
Noting again that in general we have
$\boldsymbol{x}=\hat{\boldsymbol{e}}(\hat{\boldsymbol{e}}\cdot\boldsymbol{x})-\hat{\boldsymbol{e}}\times\left(\hat{\boldsymbol{e}}\times\boldsymbol{x}\right)$, and
we can write
\[\begin{align}
\boldsymbol{v}_{ij}'-\boldsymbol{v}_{ij}&=\hat{\boldsymbol{r}}_{ij}\left(\hat{\boldsymbol{r}}_{ij}\cdot(\boldsymbol{v}_{ij}'-\boldsymbol{v}_{ij})\right) -\hat{\boldsymbol{r}}_{ij}\times\left[\hat{\boldsymbol{r}}_{ij}\times\left(\boldsymbol{v}_{ij}'-\boldsymbol{v}_{ij}\right)\right]\\
&=\hat{\boldsymbol{r}}_{ij}\left(\hat{\boldsymbol{r}}_{ij}\cdot\left(\boldsymbol{g}_{ij}'-\boldsymbol{g}_{ij}\right)\right) -\left(1+\tilde{I}^{-1}\right)^{-1}\hat{\boldsymbol{r}}_{ij}\times\left[\hat{\boldsymbol{r}}_{ij}\times\left[\boldsymbol{g}_{ij}'-\boldsymbol{g}_{ij}\right]\right]
\end{align}\]
Inserting in the coefficients of restitution, we have
\[\begin{align}
\boldsymbol{v}_{ij}'-\boldsymbol{v}_{ij}&=-(1+\varepsilon^n)\hat{\boldsymbol{r}}_{ij}\left(\hat{\boldsymbol{r}}_{ij}\cdot\boldsymbol{g}_{ij}\right) -\left(1+\tilde{I}^{-1}\right)^{-1}(\varepsilon^t-1)\hat{\boldsymbol{r}}_{ij}\times\left(\hat{\boldsymbol{r}}_{ij}\times\boldsymbol{g}_{ij}\right)
\end{align}\]
Using this we can complete the expression for the linear velocity
change:
\[\begin{align}
\boldsymbol{v}_i'-\boldsymbol{v}_i&=-m_i^{-1}\,\mu_{ij}\left((1+\varepsilon^n)
\hat{\boldsymbol{r}}_{ij}\left(\hat{\boldsymbol{r}}_{ij}\cdot\boldsymbol{g}_{ij}\right)
+\left(1+\tilde{I}^{-1}\right)^{-1}(\varepsilon^t-1)
\hat{\boldsymbol{r}}_{ij}\times\left(\hat{\boldsymbol{r}}_{ij}\times\boldsymbol{g}_{ij}\right)\right)
\end{align}\]
Inserting this into the expression for the change in angular velocity,
we have
\[\begin{align*}
\boldsymbol{\omega}_i'- \boldsymbol{\omega}_i&=- R_i^{-1}\tilde{I}_i^{-1}\left[\hat{\boldsymbol{r}}_{ij}\times\left(\boldsymbol{v}_i' -\boldsymbol{v}_i\right)\right]\\
&=
m_i^{-1}\,\mu_{ij}\,R_i^{-1}\left(1+\tilde{I}\right)^{-1}(\varepsilon^t-1)\hat{\boldsymbol{r}}_{ij}\times\left[
\hat{\boldsymbol{r}}_{ij}\times\left(\hat{\boldsymbol{r}}_{ij}\times\boldsymbol{g}_{ij}\right)\right]\\
&=
-m_i^{-1}\,\mu_{ij}\,R_i^{-1}\left(1+\tilde{I}\right)^{-1}(\varepsilon^t-1)\hat{\boldsymbol{r}}_{ij}\times\boldsymbol{g}_{ij}
\end{align*}\]
The corresponding rules for particle $j$ follow with similar
reasoning or through relabing and noting that
$\boldsymbol{g}_{ji}=-\boldsymbol{g}_{ij}$. In summary, we have
\[\begin{align*}
\boldsymbol{v}_i'-\boldsymbol{v}_i&=-\frac{\mu_{ij}}{m_i}\left((1+\varepsilon^n)
\hat{\boldsymbol{r}}_{ij}\left(\hat{\boldsymbol{r}}_{ij}\cdot\boldsymbol{g}_{ij}\right)
+\frac{\varepsilon^t-1}{1+\tilde{I}^{-1}}
\hat{\boldsymbol{r}}_{ij}\times\left(\hat{\boldsymbol{r}}_{ij}\times\boldsymbol{g}_{ij}\right)\right)\\
\boldsymbol{v}_j'-\boldsymbol{v}_j&=\frac{\mu_{ij}}{m_j}\left((1+\varepsilon^n)
\hat{\boldsymbol{r}}_{ij}\left(\hat{\boldsymbol{r}}_{ij}\cdot\boldsymbol{g}_{ij}\right)
+\frac{\varepsilon^t-1}{1+\tilde{I}^{-1}}
\hat{\boldsymbol{r}}_{ij}\times\left(\hat{\boldsymbol{r}}_{ij}\times\boldsymbol{g}_{ij}\right)\right)\\
\boldsymbol{\omega}_i'-
\boldsymbol{\omega}_i&=-\frac{\mu_{ij}(\varepsilon^t-1)}{m_i\,R_i\left(1+\tilde{I}\right)}
\hat{\boldsymbol{r}}_{ij}\times\boldsymbol{g}_{ij}
\\
\boldsymbol{\omega}_j'-
\boldsymbol{\omega}_j&=-\frac{\mu_{ij}(\varepsilon^t-1)}{m_j\,R_j\left(1+\tilde{I}\right)}\hat{\boldsymbol{r}}_{ij}\times\boldsymbol{g}_{ij}
\end{align*}\]
where $\varepsilon^n$ and $\varepsilon^t$ are the normal and
tangential coefficient of elasticity respectively. See the
derivation above for more information on the symbols and their
meaning.
Example usage
<Interaction Type="HardSphere" Diameter="1" Name="Bulk">
<IDPairRange .../>
</Interaction>
Full Tag, Subtag, and Attribute List
-
Type (attribute): Must have the
value "HardSphere" to select this Interaction type.
-
Diameter (attribute): The interaction diameter
($\sigma$) of the particle pairs corresponding to this
Interaction.
This attribute is a Property specifier with a unit
of Length (see the section on
Properties for more information).
-
Elasticity (attribute): This is an optional
attribute. The elasticity, $\varepsilon^n$, controls the dissipation of
translational energy between interacting particles. This value is
typically 1 for molecular systems and between zero and one for
granular systems. If the attribute is unset, it is equivalent
to Elasticity="1".
This attribute is a Property specifier
with Dimensionless units (see
the section on Properties for more
information).
-
TangentialElasticity (attribute): This is an
optional attribute. The tangential elasticity, $\varepsilon^t$,
controls the dissipation/generation of energy between interacting
particles in the tangential/rotational direction and is typically
in the range $[-1,1]$. If the tangential elasticity is one, there
is no tangential interaction,
whereas TangentialElasticity="-1" corresponds to a complete
reversal of the relative surface velocities. It is important to
note that setting the TangentialElasticity attribute
requires that the particles have a Species
with inertia information. If the attribute is unset then
rotational dynamics are ignored and the particles can have
any Species type.
This attribute is a Property specifier
with Dimensionless units (see
the section on Properties for more
information).
-
Name (attribute): The name of the Interaction. This
name is used to identify the Interaction in the output generated
by the dynarun command. Each Interaction must have a name which is
unique.
-
IDPairRange (tag):
This IDPairRange tag specifies the
pairs of particles which interact using this Interaction. See
the section on IDPairRanges for more
information on the format of this tag.
Type="SquareWell"
Description
The "SquareWell" Interaction implements the
square-well interaction potential, illustrated in the figure below.
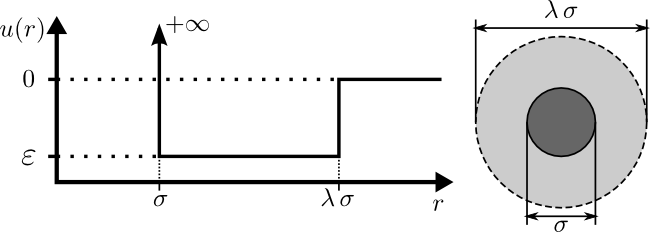
A square-well molecule is a particle which has a hard-core diameter
of $\sigma$ and is surrounded by an attractive well with a diameter
of $\lambda\,\sigma$ and a depth of $\varepsilon$. These variables
are illustrated in the diagram below:
where $u(r)$ is the interparticle potential (which is the potential
energy between two particles separated by a distance of $r$).
Square-well molecules are simple models which display the two
fundamental features of real molecules, a short range repulsion (due
to overlapping electron clouds) and longer ranged attraction (due to
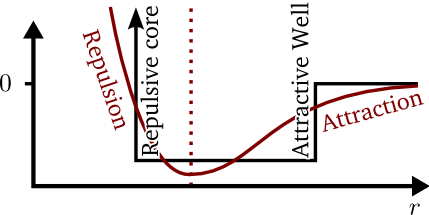
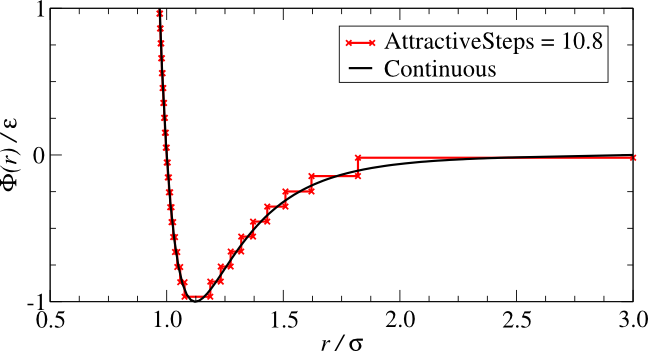
van-der-waals/London/dispersion forces). A comparison of the
square-well model (black) and a "realistic" interatomic potential
(red) is given in
the figure below:
It is clear that the square-well potential is a rough approximation
of the "realistic" potential, but its dynamics are not immediately
clear. With the "realistic" potential, it is easy to see how a pair
of particles might "fall down" the potential and attract or repulse
each other, but how does this behaviour appear in the square-well
model? When two distant square-well particles approach each other
and reach a separation of $r=\lambda\,\sigma$, they enter the well
(or "capture" each other) and a momentum impulse increases their
kinetic energy by $\varepsilon$ (they are attracted to each
other). If they then approach the inner core and reach a separation
of $r=\sigma$, they will be unable to pay the infinite energy cost
to enter the core and will instead elastically bounce off it. Once
they begin to retreat from each other (either by bouncing off the
core or by missing it) and reach a separation of
$r=\lambda\,\sigma$, they must have enough kinetic energy in the
direction of the well to escape it and pay the energy cost,
$\varepsilon$, otherwise they will bounce off the inside of the well
(both are attractive interactions).
If we used more steps to more accurately approximate the "realistic"
potential (see the "Stepped" type
Interaction), we can quite quickly capture the full behaviour of
the smooth/"realistic" potential. However, the square-well model is
so interesting because it is so simple! We can make progress in
understanding it theoretically and, if we can understand the
fundamental behaviour of square-well molecules, the fundamental
behaviour of realistic potentials can also be explained.
Example usage
<Interaction Type="SquareWell" Diameter="1" Elasticity="1" Lambda="1.5" WellDepth="1" Name="Bulk">
<IDPairRange .../>
<CaptureMap .../>
</Interaction>
Full Tag, Subtag, and Attribute List
-
Type (attribute): Must have the
value "SquareWell" to select this Interaction type.
-
Diameter (attribute): The interaction diameter
($\sigma$) of the particle pairs corresponding to this
Interaction.
This attribute is a Property specifier with a
unit of Length (see the section on
Properties for more information).
-
Elasticity (attribute): The elasticity of the
particle pairs corresponding to this Interaction. This value is
typically 1 for molecular systems and between zero and one for
granular systems.
This attribute is a Property specifier with
Dimensionless units (see the section on
Properties for more information).
-
Lambda (attribute): The well-width factor
($\lambda$) of the particle pairs corresponding to this
Interaction. Values below 1 are not valid.
This attribute is a Property specifier with
Dimensionless units (see
the section on Properties for more
information).
-
WellDepth (attribute): The interaction energy
($\varepsilon$) of the particle pairs corresponding to this
Interaction.
This attribute is a Property specifier with a unit
of Energy (see the section on
Properties for more information).
-
Name (attribute): The name of the Interaction. This
name is used to identify the Interaction in the output generated
by the dynarun command. Each Interaction must have a name which is
unique.
-
CaptureMap (tag): If present, the CaptureMap tag
should store the particle pairs which are within the well. If it
is not present, it will be automatically generated when the
configuration is next loaded by dynarun or dynamod. The data in
this tag must be correct at all times otherwise errors in the
dynamics will occur so take care when manually editing the
configuration file. See the reference entry
on CaptureMap for more details.
-
IDPairRange (tag): This IDPairRange tag specifies
the pairs of particles which interact using this
Interaction. See the section on
IDPairRanges for more information on the format of this tag.
Type="ParallelCubes"
Description
The "ParallelCubes" Interaction implements the hard cube interaction
potential where the cubes do not rotate and are axis-aligned (a
video of this system is presented to the right).
Example usage
<Interaction Type="ParallelCubes" Diameter="1" Elasticity="1" Name="Bulk">
<IDPairRange .../>
</Interaction>
Full Tag, Subtag, and Attribute List
-
Type (attribute): Must have the
value "ParallelCubes" to select this Interaction type.
-
Diameter (attribute): The interaction diameter is
the length at which the particle pairs collide with this
Interaction (the length of a cube side).
This attribute is a Property specifier with a unit of
Length (see the section on
Properties for more information).
-
Elasticity (attribute): The elasticity of the
particle pairs corresponding to this Interaction. This value is
typically 1 for molecular systems and between zero and one for
granular systems.
This attribute is a Property specifier
with Dimensionless units (see
the section on Properties for more
information).
-
Name (attribute): The name of the Interaction. This
name is used to identify the Interaction in the output generated
by the dynarun command. Each Interaction must have a name which is
unique.
-
IDPairRange (tag): This IDPairRange tag specifies
the pairs of particles which interact using this
Interaction. See the section on
IDPairRanges for more information on the format of this tag.
Type="Lines"
Description
The "Lines" Interaction implements the hard
infinitely-thin rods interaction potential (a video of this system
is presented to the right).
Example usage
<Interaction Type="Lines" Length="1" Elasticity="1" Name="Bulk">
<IDPairRange .../>
</Interaction>
Full Tag, Subtag, and Attribute List
-
Type (attribute): Must have the
value "Lines" to select this Interaction type.
-
Length (attribute): The length of the lines.
This attribute is a Property specifier with a unit
of Length (see the section on
Properties for more information).
-
Elasticity (attribute): The elasticity of the
particle pairs corresponding to this Interaction. This value is
typically 1 for molecular systems and between zero and one for
granular systems.
This attribute is a Property specifier with
Dimensionless units (see
the section on Properties for more
information).
-
Name (attribute): The name of the Interaction. This
name is used to identify the Interaction in the output generated
by the dynarun command. Each Interaction must have a name which is
unique.
-
IDPairRange (tag): This IDPairRange tag specifies
the pairs of particles which interact using this
Interaction. See the section on
IDPairRanges for more information on the format of this tag.
Type="Stepped"
Description
The "Stepped" Interaction wraps a generic spherically-symmetric
stepped Potential and uses it for
two-particle interactions. This can be used to implement many simple
systems (hard-spheres, square-wells) and many complex systems such
as a discontinuous Lennard-Jones potential. An alternative approach
is to use a "Umbrella" System event to
bind collections of particles together using
a Potential.
Example usage
Generic wrapping of a Potential.
<Interaction Type="Stepped" Name="Bulk" LengthScale="1" EnergyScale="1">
<IDPairRange .../>
<Potential .../>
<CaptureMap .../>
</Interaction>
Full Tag, Subtag, and Attribute List
-
Type (attribute): Must have the
value "Stepped" to select this Interaction type.
-
Name (attribute): The name of the Interaction. This
name is used to identify the Interaction in the output generated
by the dynarun command. Each Interaction must have a name which is
unique.
-
LengthScale (attribute): The length scale by which
the Potential is scaled for each
particle. For example, if
a Lennard-Jones type
Potential is used, this sets the $\sigma$ value for each
particle.
This attribute is a Property specifier with a unit
of Length (see the section on
Properties for more information).
-
EnergyScale (attribute): The energy scale by which
the Potential is scaled for each
particle. For example, if
a Lennard-Jones type
Potential is used, this sets the $\varepsilon$ value for each
particle.
This attribute is a Property specifier with a unit
of Length (see the section on
Properties for more information).
-
Potential (tag):
The Potential tag specifies the stepped
potential used for this interaction. Please see the section on
the Potential tag for more information.
-
CaptureMap (tag): If present, the CaptureMap tag
should store the current step of any particle pairs which are
inside the Interaction range of the potential. If it is not
present, it will be automatically generated when the configuration
is next loaded by dynarun or dynamod. The data in this tag must be
correct at all times otherwise errors in the dynamics will occur
so take care when manually editing the configuration file. See the
reference entry on CaptureMap for more
details.
-
IDPairRange (tag): This IDPairRange tag specifies
the pairs of particles which interact using this
Interaction. See the section on
IDPairRanges for more information on the format of this tag.
Type="ThinThread"
Description
The "ThinThread" Interaction implements a
square-well potential which has hysteresis. The particle behaves as
a hard sphere, but once a pair of particles collide with the hard
core, they enter the well. The energy of this well must be paid to
escape, thus the model loses energy each time particles enter the
well. Once particles escape the well, they must again collide with
the core before they can re-enter the well (as illustrated in the
figure below). This model may be used to model wet granulate and the
formation of liquid bridges as when wet granular particles contact
each other a liquid bridge stretches between them. Breaking this
bridge requires an amount of energy and distance to overcome the
surface tension (modelled by the well), but once the bridge is
broken the particles will only interact again at short distances.
Example usage
<Interaction Type="ThinThread" Diameter="1" Elasticity="1" Lambda="1.5" WellDepth="1" Name="Bulk">
<IDPairRange .../>
<CaptureMap .../>
</Interaction>
Full Tag, Subtag, and Attribute List
-
Type (attribute): Must have the
value "ThinThread" to select this Interaction type.
-
Diameter (attribute): The interaction diameter
($\sigma$) of the particle pairs corresponding to this
Interaction.
This attribute is a Property specifier with a unit
of Length (see the section on
Properties for more information).
-
Elasticity (attribute): The elasticity of the
particle pairs corresponding to this Interaction. This value is
typically 1 for molecular systems and between zero and one for
granular systems.
This attribute is a Property specifier with
Dimensionless units (see
the section on Properties for more
information).
-
Lambda (attribute): The well-width factor
($\lambda$) of the particle pairs corresponding to this
Interaction. Values below 1 are not valid.
This attribute is a Property specifier with
Dimensionless units (see
the section on Properties for more
information).
-
WellDepth (attribute): The interaction energy
($\varepsilon$) of the particle pairs corresponding to this
Interaction.
This attribute is a Property specifier with a unit
of Energy (see the section on
Properties for more information).
-
Name (attribute): The name of the Interaction. This
name is used to identify the Interaction in the output generated
by the dynarun command. Each Interaction must have a name which is
unique.
-
CaptureMap (tag): If present, the CaptureMap tag
should store the particle pairs which are within the well. If it
is not present, it will be automatically generated when the
configuration is next loaded by dynarun or dynamod. The data in
this tag must be correct at all times otherwise errors in the
dynamics will occur so take care when manually editing the
configuration file. See the reference entry
on CaptureMap for more details.
-
IDPairRange (tag): This IDPairRange tag specifies
the pairs of particles which interact using this
Interaction. See the section on
IDPairRanges for more information on the format of this tag.
Type="SquareBond"
Description
The "SquareBond" Interaction implements a
square-well potential with an infinite interaction energy. This
allows you to bond particles together to form polymeric structures.
Example usage
<Interaction Type="SquareBond" Diameter="1" Elasticity="1" Lambda="1.5" Name="Bulk">
<IDPairRange .../>
</Interaction>
Full Tag, Subtag, and Attribute List
-
Type (attribute): Must have the
value "SquareBond" to select this Interaction type.
-
Diameter (attribute): The interaction diameter
($\sigma$) of the particle pairs corresponding to this
Interaction.
This attribute is a Property specifier with a unit
of Length (see the section on
Properties for more information).
-
Elasticity (attribute): The elasticity of the
particle pairs corresponding to this Interaction. This value is
typically 1 for molecular systems and between zero and one for
granular systems.
This attribute is a Property specifier with
Dimensionless units (see
the section on Properties for more
information).
-
Lambda (attribute): The bond-width factor
($\lambda$) of the particle pairs corresponding to this
Interaction. Values below 1 are not valid.
This attribute is a Property specifier with
Dimensionless units (see
the section on Properties for more
information).
-
Name (attribute): The name of the Interaction. This
name is used to identify the Interaction in the output generated
by the dynarun command. Each Interaction must have a name which is
unique.
-
IDPairRange (tag): This IDPairRange tag specifies
the pairs of particles which interact using this
Interaction. See the section on
IDPairRanges for more information on the format of this tag.
CaptureMap
CaptureMaps are used by DynamO to track which particles are
currently interacting with other particles using that
Interaction. For example,
the SquareWell
type Interactions uses the capture map to record all
pairs of particles who are inside each others attractive well. This
information must be saved and loaded with the configuration file as,
if a particle is on the edge of the well, it is impossible to
determine if they are captured or not from their position
alone. Without using a CaptureMap, this ambiguity would lead to the
simulation either losing or gaining energy after a save/load.
If DynamO does not see a CaptureMap in the configuration
file, but the Interaction requires one it will automatically
calculate the CaptureMap from the particle positions.
Whenever we change a configuration file by hand, its very likely
that we will invalidate any data stored within CaptureMap
tags inside the file. The simplest way of correcting this error is
to delete the CaptureMap tags. This forces DynamO to rebuild
them when it next loads the configuration file. You should note that
deleting the CaptureMap might cause the potential energy of the
system to change slightly, so it should be avoided if energy
conservation is desired.
Local
Locals are sources of events for particles where each event only
involves a single particle and this event only occurs when the
particle is within certain regions of the simulation. The standard
example of a Local is a wall or some other boundary of the
simulation.
Type="Wall"
Description
The "Wall" Local implements an infinite
plane/wall. This Local is typically used as a boundary of the
simulation and may also be "thermalised" to inject or remove energy
from the system. As these planes/walls are infinite, you must take
care that they are axis-aligned if the system is periodic. They are
not compatible with shearing simulations
(see Lees-Edwards boundary conditions) unless the
normal is aligned with the $y$-axis (which would defeat the purpose
of using Lees-Edwards boundaries).
Example usage
<Local Type="Wall" Name="GroundPlate" Elasticity="1" Diameter="1" Temperature="0.5">
<IDRange Type="All"/>
<Norm x="0" y="1" z="0"/>
<Origin x="0" y="-7.5" z="0"/>
</Local>
Full Tag, Subtag, and Attribute List
-
Type (attribute): Must have the
value "Wall" to select this Interaction type.
-
Diameter (attribute): The diameter of the
particles which interact with this wall. Although the diameter
must be given here (to be consistent with Interactions), you
should note that particles actually collide with the wall when
they reach a separation equal to half the diameter.
This attribute is a Property specifier with a unit
of Length (see the section on
Properties for more information).
-
Elasticity (attribute): The elasticity of the
particle collisions. This value is typically 1 for molecular
systems and between zero and one for granular systems.
This attribute is a Property specifier with
Dimensionless units (see
the section on Properties for more
information).
-
Temperature (attribute): The temperature is an
optional tag. If it is present, whenever a particle hits the
wall, the normal component of its velocity will be randomly
reassigned from a distribution with the specified
temperature. If it is not present, the particles will collide
according to the elasticity.
-
Name (attribute): The name of the Local. This name
is used to identify the Local in the output generated by the
dynarun command. Each Local must have a name which is
unique.
-
IDRange (tag): This IDRange tag specifies the
particles which interact using this Local. See
the section on IDRanges for more
information on the format of this tag.
-
Norm (tag): The normal of the plane of the
wall. The value is renormalised by DynamO on load.
-
x (attribute): The $x$-component of the normal.
-
y (attribute): The $y$-component of the normal.
-
z (attribute): The $z$-component of the normal.
-
Origin (tag): A point on the plane of the
wall.
-
x (attribute): The $x$-coordinate of the point.
-
y (attribute): The $y$-coordinate of the point.
-
z (attribute): The $z$-coordinate of the point.
Global
Globals, like Locals, specify events which only affect a single
particle in the system. The difference between Globals and Locals is
that a Global event will occur to a particle, regardless of its
location in the system. The most common type of Global used is the
cellular neighbour list.
Type="Cells"
Description
The "Cells" Global implements a cellular
neighbour list, which may be used by the Scheduler to optimise the
simulation. The neighbourlist will track all particles that match
its IDRange and can provide information on which tracked particles
are within the neighbourhood of other particles or points.
Example usage
<Global Type="Cells" Name="SchedulerNBList" NeighbourhoodRange="1">
<IDRange .../>
</Global>
Full Tag, Subtag, and Attribute List
-
Type (attribute): Must have the
value "Cells" to select this Global type.
-
Name (attribute): The name of the Global. This
name is used to identify the Global in the output generated by
the dynarun command. Each Global must have a name which is
unique. In particular, if this cellular neighbour list is to be
used by the "NeighbourList" type Scheduler, it should have the
name "SchedulerNBList".
-
NeighbourhoodRange (attribute): This optional
attribute sets the minimum distance at which particles are
considered to be in the neighbourhood of other particles or
points. If the neighbour list is used by
the "NeighbourList" type
Scheduler, this distance should be at least equal to or
greater than the maximum Interaction distance in the system. If
this tag is unset, the maximum interaction distance is
automatically calculated.
-
IDRange (tag): This IDRange tag specifies the
particles which are tracked in the cellular neighbour list. See
the section on IDRanges for more
information on the format of this tag.
System
System tags represent events which are unique. There is only one
event time generated by a System, and this event may alter zero or
many particles at once. If an event type does not fit into the
categories of
Global, Local,
or Interaction, it will be implemented
via a System event. The most common type of system event is the
Andersen thermostat.
Type="Andersen"
Description
The "Andersen" System implements an Andersen
thermostat. An Andersen thermostat functions by randomly reassigning
the velocities of individual particles from a Gaussian distribution
with a specified temperature. These reassignments occur at random
time intervals given by a Poisson distribution with a specified mean
free time.
This thermostat adds thermal "noise" into the system, and so
dynamical properties will be affected by its action. To avoid
completely losing the dynamics of the system, the thermostat is
usually controlled to ensure that only a certain fraction of the
total events of the system are thermostat events. In DynamO, this
fraction is specified by a
SetPoint attribute. Every SetFrequency events, the
mean free time of the thermostat is adjusted to try to match
the SetPoint.
If you want the mean free time to remain fixed during a simulation
and want to disable the frequency control please ensure that you do
not define the SetFrequency or
SetPoint attributes. If either attribute is missing the
frequency control will be disabled.
Example usage
<System Type="Andersen" Name="Thermostat" MFT="1.0" Temperature="1.0" SetPoint="0.05" SetFrequency="100">
<IDRange Type="All"/>
</System>
Full Tag, Subtag, and Attribute List
-
Type (attribute): Must have the
value "Andersen" to select this System type.
-
Name (attribute): The name of the System event. This
name is used to identify the System in the output generated by the
dynarun command. Each System must have a name which is unique.
-
MFT (attribute): The current mean free time over
which the thermostat is applied. Please note, this is the
mean free time of the thermostat not the mean free time
per particle.
-
Temperature (attribute): The temperature ($k_B\,T$)
of the thermostat.
-
SetPoint (attribute): This attribute is optional and
only takes effect if the SetFrequency attribute is also
defined. The target fraction of events which should be
applications of the thermostat. This is effectively the damping
constant of the thermostat.
-
SetFrequency (attribute): This attribute is optional
and only takes effect if the SetPoint attribute is also
defined. The thermostat mean free time is adjusted every
SetFrequency events to attempt to match the SetPoint fraction of
events.
-
Dimensions (attribute): A number indicating how many
dimensions of the particles are to be thermostatted. The default
is all (=3). If set to 2, the $z$ dimension is not affected by the
thermostat, and if set to 1, neither the $y$ or $z$-dimensions are
affected.
-
IDRange (tag): This IDRange tag specifies the
particles to which the thermostat is applied. See
the section on IDRanges for more
information on the format of this tag.
Type="Umbrella"
Description
The "Umbrella" System implements an umbrella potential, allowing
a Potential to be specified between the
centres of mass of two collections of particles.
Example usage
An umbrella potential between two groups of 64 particles:
<System Type="Umbrella" Name="UmbrellaPotential" LengthScale="1" EnergyScale="1">
<IDRange Type="Ranged" Start="0" End="63"/>
<IDRange Type="Ranged" Start="64" End="127"/>
<Potential Type="Stepped" Direction="Right">
<Step R="0.25" E="0.25"/>
<Step R="0.5" E="0.5"/>
<Step R="0.75" E="0.75"/>
<Step R="1.0" E="1.0"/>
<Step R="1.25" E="1.25"/>
<Step R="1.5" E="1.5"/>
<Step R="1.75" E="1.75"/>
<Step R="2.0" E="2.0"/>
<Step R="2.25" E="2.25"/>
<Step R="2.5" E="2.5"/>
<Step R="2.75" E="2.75"/>
<Step R="3.0" E="3.0"/>
<Step R="3.25" E="3.25"/>
<Step R="3.5" E="3.5"/>
<Step R="3.75" E="3.75"/>
<Step R="4.0" E="4.0"/>
<Step R="4.5" E="4.5"/>
<Step R="5.0" E="5.0"/>
<Step R="5.5" E="5.5"/>
<Step R="6.0" E="6.0"/>
</Potential>
</System>
Full Tag, Subtag, and Attribute List
-
Type (attribute): Must have the
value "Umbrella" to select this System type.
-
Name (attribute): The name of the System event. This
name is used to identify the System in the output generated by the
dynarun command. Each System must have a name which is unique.
-
LengthScale (attribute): The length scale by which
the Potential is scaled by. For example,
if a Lennard-Jones type
Potential is used, this sets the $\sigma$ value.
-
EnergyScale (attribute): The energy scale by which
the Potential is scaled by. For example,
if a Lennard-Jones type
Potential is used, this sets the $\varepsilon$ value.
-
IDRange (tag$\times$2): There
are two IDRange tags which specify the two
ranges of particles which interact using the umbrella
potential. See the section on IDRanges for
more information on the format of these tags.
-
Potential (tag):
The Potential tag specifies the stepped
potential used for the umbrella potential. Please see the section
on the Potential tag for more
information.
Dynamics
The Dynamics tag specifies the equations of motion of the
system. The standard variant is the "Newtonian" type, but there are
other types available which allow the addition of an external force
such as gravity.
Type="Newtonian"
Description
The "Newtonian" Dynamics type is the standard
Dynamics implementation in DynamO. All particles are moving under
standard Newtonian dynamics, without the influence of external
forces.
Example usage
<Dynamics Type="Newtonian"/>
Full Tag, Subtag, and Attribute List
-
Type (attribute): Must have the
value "Newtonian" to select this Dynamics type.
Type="NewtonianGravity"
Description
The "NewtonianGravity" Dynamics type allows an
external acceleration to be included in the dynamics.
Example usage
<Dynamics Type="NewtonianGravity" ElasticV="1.0">
<g x="0" y="-1" z="0"/>
</Dynamics>
Full Tag, Subtag, and Attribute List
-
Type (attribute): Must have the
value "NewtonianGravity" to select this Dynamics type.
-
ElasticV (attribute): An optional tag. If this tag
is present, this tag specifies the velocity below which particle
interactions will be turned elastic (used to prevent inelastic
collapse).
-
tc (attribute): An optional tag. If this tag is
present, this tag specifies the minimum re-collision rate which
turns an interaction elastic (used to prevent inelastic
collapse). For more information
see this
paper
(also available on
arxiv).
-
g (tag): A tag specifying the vector of the external
acceleration.
-
x, y, z, (attributes): The
components of the external acceleration.
Type="NewtonianMC"
Description
The "NewtonianMC" Dynamics implements a
multicanonical simulation. Multicanonical simulations deform the
energy potential of the system to accelerate the dynamics. A
descriptive paper on the technique is
"Multicanonical
Ensemble Generated by Molecular Dynamics Simulation for Enhanced
Conformational Sampling of Peptides". The following notes will
be based around the notation of this paper.
If we are in the canonical ensemble, the probability of being in a
certain configurational energy, $E$, is
\[P_c(E)=\frac{1}{Z_{c}} n(E) \exp\left[-E/k_BT\right] \]
But for efficient sampling of all energies we would prefer it if the
probability of each energy is constant and equal.
\[ P_{mc} (E) = \frac{1}{Z_{mc}} n(E) \exp\left[-E/k_BT-W(E)\right] = \textrm{constant} \]
This is the ideal multi-canonical simulation, as we sample all
energies equally. However, to perform the ideal multi-canonical
simulation we need to know the $W(E)$ function.
\[ W(E) = \ln n(E) - E / k_B T = \ln P_c(E) \]
This requires knowing the density of states ($n(E)$), but if these
quantities are known, the problem of sampling the system is already
solved. So what we need is an iterative way to determine the $W(E)$
function. The simplest technique is to run a simulation with
$W^{(0)}(E)=0$, and then iterate towards the optimal weighting with
this expression:
\[ W^{(i+1)}(E) = W^{(i)}(E) + \ln P^{(i)}_{mc}(E) \]
There are more advanced techniques available in the literature which
have been implemented and these are available as scripts which are
run outside of DynamO. These are discussed elsewhere and only the
DynamO interface is described here.
It should be noted that the $W$ function is effectively an
additional potential in the Hamiltonian of the system:
\[ P_{mc} (E) = \frac{1}{Z_{mc}} n(E) \exp\left[-(E+E_{MC}(E))/k_B\,T\right] \]
where $E_{MC}(E) = k_B\,T\,W(E)$ is the multicanonical potential.
If we want to specify a multicanonical potential (or $W$ function)
in DynamO, we must do so using a stepped function. Each step has the
same width and is centered around an energy value. The width of all
steps is set using the EnergyStep attribute of
the PotentialDeformation tag. The energy and $W$ value of
each step is specified in a list of W tags inside
the PotentialDeformation tag. If there is no step specified
for an energy, its $W$ value is automatically assumed to be zero.
Example usage
<Dynamics Type="NewtonianMC">
<PotentialDeformation EnergyStep="0.01">
<W Energy="0" Value="-8.16128284222000e+02"/>
<W Energy="-18" Value="-5.39112459746000e+02"/>
...
</PotentialDeformation>
</Dynamics>
Full Tag, Subtag, and Attribute List
-
Type (attribute): Must have the
value "NewtonianMC" to select this Dynamics type.
-
PotentialDeformation (tag): This tag encloses the
stepped deformation potential used in the multicanonical
simulation.
-
EnergyStep (attribute): The width of each
deformation potential step.
-
W (tag): This tag contains the location of the
deformation potential step and its $W$ value.
-
Energy (attribute): The centre energy of the
deformation energy step.
-
Value (attribute): The $W$ value of the
deformation energy step.
Property
Property tags are a mechanism which allows you to specify large
amounts of information which may or may not vary on a per-particle
basis. For example, if every Particle in the system is
a HardSphere type Interaction with the
same diameter of 1.5, you might use the following Interaction:
<Interaction Type="HardSphere" Diameter="1.5" Elasticity="1" Name="Bulk">
<IDPairRange .../>
</Interaction>
The values of the Diameter and Elasticity are called
a numeric properties where the value of the property
specifier is the value of the property. But what if you want a
polydisperse system, where each particle may have a unique mass and
diameter? In this case we would use named Properties:
<Interaction Type="HardSphere" Diameter="D" Elasticity="1" Name="Bulk">
<IDPairRange .../>
</Interaction>
Here we've used the name "D" to refer to a new named
Property. Whenever DynamO encounters a property specifier, such as
the Diameter attribute above, and fails to convert it directly into
a floating-point number due to the presence of letters in its name,
it assumes that the property is a named property. This named
property can also be reused in other property specifiers at the same
time, such as in a Wall type Local:
<Local Type="Wall" Name="GroundPlate" Elasticity="1" Diameter="D">
...
</Local>
And now both the Wall Local and the HardSphere Interaction will use
the same diameter for each particle. Each named Property must be
defined in the Properties tag in the configuration file. For
example, if we wanted to define the mass and diameter of each
particle individually, we would define two "PerParticle" Properties
like so:
<Properties>
<Property Type="PerParticle" Name="D" Units="Length"/>
<Property Type="PerParticle" Name="M" Units="Mass"/>
</Properties>
You should note that the units of the Property must correspond to
the units of the property specifier. If you check
the HardSphere Interaction
documentation, you can confirm that the Diameter attribute has units
of Length (The available units
include Dimensionless, Length, Area, Volume, Time, Mass,
and Energy).
We can use a named property in the Species
definition to use this new per-particle mass:
<Species Mass="M" Name="Bulk" Type="Point">
...
</Species>
Once the per-particle Property has been defined and referred to in
other parts of the configuration file, you must specify the value of
the property for each particle. This is done by adding an attribute
to the Pt (particle) tags with the same name as
the property. For example:
<Pt ID="0" M="1.11" D="0.323451">
<P .../>
<V .../>
</Pt>
At the moment, there is only the PerParticle type of named property,
and every single particle must have the corresponding property
attribute (in the example above, each Pt tag must have
a D and M attribute).
Pt (Particle)
Description
A Pt or Particle tag represents the
unique data of a single particle. Each particle must have at least a
position and velocity tag, but it may also include additional
attributes and tags corresponding
to Properties.
Example usage
<Pt ID="0">
<P x="1.71513720091304e+00" y="5.49987913872954e+00" z="4.32598642635552e+00"/>
<V x="1.51174422678297e+00" y="-8.06881217863154e-01" z="-8.11332120569972e-01"/>
</Pt>
Full Tag, Subtag, and Attribute List
-
ID (attribute): DynamO loads and assigns ID's to
particles in the order in which they appear in the configuration
file. This tag is therefore not read by DynamO, but is provided
in generated configuration files to make it easy to identify
particles.
-
P (tag): This tag contains the position of the
particle. In systems with periodic boundary conditions, the
dynamod/dynarun commands will output the position of the
particle image which is in the primary image (the "wrapped"
particle position). This behaviour may be disabled using
the --unwrapped option of the dynamod and dynarun
commands (the particle positions correspond to the initial
particle image's final location).
-
x (attribute): The particles $x$-coordinate.
-
y (attribute): The particles $y$-coordinate.
-
z (attribute): The particles $z$-coordinate.
-
V (tag): This tag contains the velocity of the
particle.
-
x (attribute): The $x$-component of the particle velocity.
-
y (attribute): The $y$-component of the particle velocity.
-
z (attribute): The $z$-component of the particle velocity.
Potential
The Potential tag represents a collection of discontinuities and
energies which make up a stepped potential. The location of these
steps may be manually entered using
the "Stepped" type or
automatically generated, such as in
the Lennard-Jones type.
Type="Stepped"
Description
This Potential type allows a stepped potential to be manual entered
and is the most general stepped potential available. There are two
classes of stepped potential, Left or Right, depending on which
"side" of the discontinuity is specified by the discontinuity's
energy. An illustration is given below:
In both potentials, there is an implicit step (the zero step) which
has an energy of zero. Left stepped potentials are best used for
forces which decay with $r$, whereas Right stepped potentials are
often used for forces which increase with $r$. This is for
efficiency due to two factors: The step number for a pair of
particles is not stored in memory if it has a value of zero and, for
automatically generated potentials, steps are computed from step 0
upwards (as required). Therefore, a stepped Lennard-Jones potential
is most efficiently implemented as a Left potential as most possible
particle pairs are not interacting and will be within the zero
step. A computed bond (constraining) spring potential is most
efficiently implemented using a Right potential.
Example usage
An implementation of the sixth hand stepped approximation of the
Lennard-Jones potential reported
by Chapela
et al. (1989):
<Potential Type="Stepped" Direction="Left">
<Step R="2.3" E="-0.06"/>
<Step R="1.75" E="-0.22"/>
<Step R="1.45" E="-0.55"/>
<Step R="1.25" E="-0.98"/>
<Step R="1.05" E="-0.47"/>
<Step R="1.0" E="0.76"/>
<Step R="0.95" E="3.81"/>
<Step R="0.9" E="10.95"/>
<Step R="0.85" E="27.55"/>
<Step R="0.80" E="66.74"/>
<Step R="0.75" E="1.0e+300"/>
</Potential>
Full Tag, Subtag, and Attribute List
-
Type (attribute): Must have the
value "Stepped" to select this Potential type.
-
Direction (attribute): This sets the direction of
the stepping.
For example, for potentials which have zero
interaction energy at long distances, the "Left" direction is the
most convenient. The Lennard-Jones potential is an example of such
a potential. In this case, each Step tag specifies a
location of a discontinuity and the energy on its left hand side
($r^-$). The outermost step is assumed to have an energy of zero.
A spring potential is an example where the "Right" direction
is most convenient as it has zero interaction energy at $r=0$. In
this case, each Step tag specifies a location of a
discontinuity and the energy on its right hand side ($r^+$). The
innermost step has a energy of zero
-
Step (tag): This tag represents a single
discontinuity/step of the stepped potential. On load these steps
are sorted by their R value and allocated numbers as outlined above.
-
R (attribute): The radial separation of the
discontinuity.
-
E (attribute): The energy of the potential on
either the left or right hand side of the discontinuity
(depending on the Direction).
Type="LennardJones"
Description
This Potential is a stepped approximation to the truncated and
shifted Lennard-Jones potential. The approximation and its
optimisation is reported in the
paper: "Mapping
continuous potentials to discrete forms," by Thomson, Lue, and
Bannerman. This paper should be consulted for a more detailed
discussion of the parameters below. An example of a accurate
approximation is given below:
The set-up of this stepped potential is outlined below.
Example usage
<Potential Type="LennardJones" Sigma="1" Epsilon="1" CutOff="3" AttractiveSteps="10.8"/>
Full Tag, Subtag, and Attribute List
-
Type (attribute): Must have the
value "LennardJones" to select this Potential type.
-
Sigma (attribute): Sets the distance $\sigma$ at
which the interaction energy is zero.
-
Epsilon (attribute): Sets the energy $\varepsilon$
of the attractive well.
-
Cutoff (attribute): The cutoff radius of the
potential.
-
AttractiveSteps (attribute): The order of
approximation of the potential. Setting AttractiveSteps="10.8" is
sufficient for a close reproduction of the thermodynamic
properties provided the default UMode and RMode values are used.
-
RMode (attribute, optional): Select the method used
to calculate the position of each step. This may be "DeltaR",
"DeltaU", or "DeltaV". If this attribute is not set, the
default/recommended value is used (currently "DeltaU").
-
UMode (attribute, optional): Select the method used
to calculate the energy of each step. This may be "Left", "Right",
"Midpoint", "MidVolume", "Virial", or "Volume". If this attribute
is not set, the default/recommended value is used (currently
"Volume"). If "Virial" is selected, the Temperature
attribute must also be set.
-
Temperature (attribute, optional): If
the UMode is set to "Virial" you must provide the
temperature used for the approximation in this attribute.
IDRange
IDRanges are used to specify a range
of Particle IDs. These are used in
Species, Local, Global
and Structure objects to specify the set of
particles to which they apply.
Type="All"
Description
An ID Range which maps to all particles in the
simulation.
Example usage
<IDRange Type="All"/>
Full Tag, Subtag, and Attribute List
-
Type (attribute): Must have the value "All"
to select this range type.
Type="None"
Description
An ID Range which does not map to any particles.
Example usage
<IDRange Type="None"/>
Full Tag, Subtag, and Attribute List
-
Type (attribute): Must have the
value "None" to select this range type.
Type="Ranged"
Description
An ID Range which maps to a sequential list of
particles simulation. The range is inclusive (both its start and end
points are inside the range).
Example usage
The following example IDRange includes the particle IDs 0, 1,
2, 3, 4, and 5.
<IDRange Type="Ranged" Start="0" End="5"/>
Full Tag, Subtag, and Attribute List
-
Type (attribute): Must have the
value "Ranged" to select this range type.
-
Start (attribute): The lowest value ID which is
inside the range.
-
End (attribute): The highest value ID which is
inside the range.
Type="List"
Description
An ID Range which maps to a specified list of
particles. This is the most general type of Range possible, as all
IDs inside the range must be specfied individually.
Example usage
The following example IDRange includes the particle IDs
0, 2, and 5.
<IDRange Type="List">
<ID val="0"/>
<ID val="2"/>
<ID val="5"/>
</IDRange>
Full Tag, Subtag, and Attribute List
-
Type (attribute): Must have the
value "List" to select this range type.
-
ID (tag): A tag containing an ID/entry in the
list.
-
val (attribute): The ID of the entry.
Type="Union"
Description
An IDRange which combines several IDRanges into
one.
Example usage
<IDRange Type="Union">
<IDRange .../>
<IDRange .../>
...
</IDRange>
Full Tag, Subtag, and Attribute List
-
Type (attribute): Must have the
value "Union" to select this range type.
-
IDRange (tag): A list of IDrange subtags which
describe the IDRanges to be combined.
IDPairRange
IDPairRanges are used to specify the pairs of particles to
which an Interaction applies. Each IDPairRange represent a
collection or set of ID pairs.
Type="All"
Description
An ID Range which maps to all pairs of particles
in the simulation.
Example usage
<IDPairRange Type="All"/>
Full Tag, Subtag, and Attribute List
-
Type (attribute): Must have the value "All"
to select this range type.
Type="None"
Description
An ID Range which doesn't map to any pair of
particles.
Example usage
<IDPairRange Type="None"/>
Full Tag, Subtag, and Attribute List
-
Type (attribute): Must have the
value "None" to select this range type.
Type="Pair"
Description
This creates a IDPairRange from two
IDRanges. Each particle ID in the first range is paired with all of
the particle IDs in the second range. Pairs of particle IDs which
only match one range are not included in the pair range.
Example usage
This example pairs particle IDs 0,1,2 with IDs 3,4,5. Pairs only
within one range, such as (0,1) or (3,4), are not included. All
IDRangePairs are symmetric, so both (1,4) and (4,1) are included in
this example range.
<IDPairRange Type="Pair">
<IDRange Type="Ranged" Start="0" End="2"/>
<IDRange Type="Ranged" Start="3" End="5"/>
</IDPairRange>
Full Tag, Subtag, and Attribute List
-
Type (attribute): Must have the
value "Pair" to select this range type.
-
IDRange ($2\times$tag): There must be two IDRange tags
present and each specifies one of the IDRanges used to make the
IDPairRange. The order in which the IDRanges are specifed is
unimportant. See the section on the available
IDRange types for more information.
Type="Single"
Description
This creates a IDPairRange from one
IDRange. Every particle ID in the IDRange is paired with every other
particle in the IDRange.
Example usage
This example pairs particle IDs 0-0, 0-1, 0-2, 1-1, 1-2, and 2-2.
<IDPairRange Type="Single">
<IDRange Type="Ranged" Start="0" End="2"/>
</IDPairRange>
Full Tag, Subtag, and Attribute List
Type="Self"
Description
This creates a IDPairRange from one
IDRange. Every particle ID in the IDRange is paired with itself.
Example usage
This example pairs particle IDs 0-0, 1-1, and 2-2.
<IDPairRange Type="Self">
<IDRange Type="Ranged" Start="0" End="2"/>
</IDPairRange>
Full Tag, Subtag, and Attribute List
Type="List"
Description
This is the most versatile IDPairRange available
as it allows you to specify the particle pairings individually.
Example usage
This example matches the particle pairings (0,1), (1,2), and
(2,3). As IDPairRanges are symmetric, the pairings (1,0), (2,1), and
(3,2) are also included. This example might be used to bond
particles together into a chain.
<IDPairRange Type="List">
<IDPair ID1="0" ID2="1"/>
<IDPair ID1="1" ID2="2"/>
<IDPair ID1="2" ID2="3"/>
</IDPairRange>
Full Tag, Subtag, and Attribute List
-
Type (attribute): Must have the
value "List" to select this range type.
-
IDPair (tag): Each IDPair tag specifies one
pairing of particle IDs.
-
ID1 (attr): The first ID of the pair. The
order of the IDs is unimportant and DynamO always writes the
configuration out so that ID1 is the lowest of the pair.
-
ID2 (attr): The second ID of the pair.
Type="Union"
Description
This creates a IDPairRange from a
combination/union of several IDPairRanges.
Example usage
<IDPairRange Type="Union">
<IDPairRange .../>
<IDPairRange .../>
...
</IDPairRange>
Full Tag, Subtag, and Attribute List
-
Type (attribute): Must have the
value "Union" to select this range type.
-
IDPairRange (tag): Each IDPairRange sub tag
represents one of the IDPairRanges to be combined.
Type="Chains"
Description
This IDPairRange is provided as a convenient way
to bond sequential particle IDs into linear chains.
Example usage
If the example below was used with
a square-bond Interaction, it would
bond a set of particles together into chains of 4.
This example matches the particle pairings (0,1), (1,2), (2,3) and
(4,5), (5,6), (6,7). As IDPairRanges are symmetric, the pairings
(1,0), (2,1), etc. are also included.
<IDPairRange Type="Chains" Start="0" End="7" Interval="4"/>
Full Tag, Subtag, and Attribute List
-
Type (attribute): Must have the
value "Chains" to select this range type.
-
Start (attribute): The first ID of the sequential
particle IDs.
-
End (attribute): The last ID of the sequential
particle IDs.
-
Interval (attribute): How long the chains
are. Please note that End - Start must be a whole multiple of
Interval.
Page last modified: Thursday 29th July 2021